To view the plugin source code, please visit the plugin’s GitHub repository.
 Click to play the movie on Youtube. This movie shows the first application of groupSIFT in Loo et al. (2019) NeuroImage: Clinical
Click to play the movie on Youtube. This movie shows the first application of groupSIFT in Loo et al. (2019) NeuroImage: Clinical
The GROUPSIFT EEGLAB Plugin
This page is for those who wants to cooperate with me to test groupSIFT toolbox and EEGLAB plugin. Unfortunately, at this point we cannot provide much user support. We know this page does not cover full detail either (including how to determine SIFT parameters). So this plugin is for advanced user who have good experience with Matlab and SIFT. When you use it, please do so at your own risk!
If you are interested in learning basic SIFT functions using its own built-in simulator, see this page
Environment
- I originally developed it with Matlab R2013a runninig on Fedora 22 (64bit) with dual monitors with 1600 x 1200 resolution. In a recent project, I updated it for Matlab 2017b, EEGLAB 14.1.2, and SIFT 1.52 (with customized movie function). I have not fully investigated dependency on different environments other than Linux. Matlab Image Processing Toolbox is necessary to use bwlabel().
Required preprocessing
- You need EEGLAB .set files that are processed with ICA and subsequent IC selection: ALL the ICs in your data will go to SIFT preprocess. You don’t want to use 100 ICs that will create 100 x 100 x frequency x time tensor for n subjects in the end! There is also datapont-to-parameter ratio you want to consider. Also, DIPFIT must be done. Use ICLabel() plugin to select ICs with ‘brain’ label probability > 0.7, for example.
- Downsample the data to nearly 100-120 Hz using the additional two options in this way: pop_resample(EEG, 100, 0.8, 0.4) The extra optional parameters are to use mild low-pass filter slope in anti-aliasing to suppress AR model order. See this page for detail.
- Separate conditions so that one .set file, one condition. All the SIFT-related additional preprocessing should be applied to each of subdivided single-condition data sets separately.
- Note that groupSIFT is independent of EEGLAB STUDY.
- Follow this rule in using the groupSIFT all the time: Locate the preprocessed .set files into a dedicated folder which does not have anything other than the processed files. For example, if you have condition A, condition B, as well as you plan to test A-B, create folders for A, B, and A-B separately.
- Also, it is recommended to move to the working folder every time you step forward. groupSIFT interactive file loader in GUI finds the local files with the extension filter.
- Do not use ‘_’ in the file name. This character needs to be preserved to identify prefix words.
groupSIFT GUI menu explained
1.Run SIFT Batch
Click this item to perform SIFT on the selected multiple .set files by batch mode. Again, make sure that this is applied for all the individual, condition-separated .set files. If you have two conditions, you have to go through this processes separately for each condition. rPDC and dDTF08 are computed because these are the ones you can justify the use in papers. rPDC is theoretically better, but it has known bug/problem in the highest frequency result. dDTF08 is widely used. rPDC’s result has broader spreading in the time-frequency domain, while dDTF’s result has a sharper resolution in the time-domain. Let GUI windows stay while a process goes on. Closing it in the middle will crash the process.
SIFT tips (08/15/2020 added)
I found that the equation used in calculating datapoint-to-parameter ratio is incorrect. According to Schlögl and Supp (2006) as well as Korzeniewska et al. (2008), the equation must be (num_IC*model_order)/window_length*num_trials. This change affects how you determine parameters, in a ‘better’ way I would say as shown below: 1) more ICs can be included; 2) less number of trials can be tolerated; 3) shorter sliding window can be used. This change will particularly impacts continuous data analysis, as the current equation would probably allow sliding window length of a few minutes! In fact, this datapoint-to-parameter ratio has been a limiting factor for me to apply SIFT with confidence.
To obtain the corrected datapoint-to-parameter ratio based on the above-suggested reason, make the following change on est_checkMVARParams() line 85
%winrat = ((M^2)*p)/(wlen*T);
winrat = ((M)*p)/(wlen*T);
That being said, when I discussed this issue with the author, he also told me that if we make a correction, the estimate would be overly lax and would even become useless. I kind of see the point from my experience. Somehow, most of SIFT validation measures are always either too lax (the stability test) or too stringent (the whiteness tests), which are generally hard to follow. In conclusion, I would recommend the above fix to enjoy more degrees of freedom in the analysis design, while trying to stay as conservative (i.e., lower the number, more conservative!) as possible.
Note also that renormalized partial directed coherence (rPDC) always has noise near the highest frequency. I confirmed it with the original author of SIFT, Dr. Tim Mullen. It is advised that one always excludes near-highest freq results in rPDC.
2.Varidate AR models
Check the summary plots and consider to remove outlier subjects (horizontal axis represents set file indices). Check the group-mean value (the rightmost bar) of the datapoint-to-parameter ratio. to guarantee validity of the AR modeling stage. By the way, the definition of datapoint-to-parameter ratio is calculated differently from the original paper, so be careful. For detail, see this page.
3.Convert to group anatomical ROIs
FWHM determines the smoothing width for the dipole density. Typically, fMRI==8mm, PET==20mm. You also preselect the minimum number of subjects so that hereafter you focus on anatomical regions with majority of subjects (e.g., 80%) contributes non-zero dipole density. Press the ‘Compute upper bound for estimation’ button to confirm the result. If you are satisfied with the estimation, enter the file prefix name (again, DO NOT use ‘_’) and press the button in the bottom ‘Select ALL .set files and START’.
4.Compute t-scores & p-values
Specify the folder that has the precomputed results from the process above. To test the difference A-B, specify the folders for A and B, and further specify the new folder to save the A-B results. Uncorrected p-value here determines the size of the pixel clusters in the later time-frequency plots. The number of iteration should not be less than 2000 because the surrogate distribution is used for nonparametric tests which requires good tail support. For multiple comparison correction, weak family-wise error rate correction (FWER) is applied to perform cluster-level correction. The mass of cluster, sum of t-scores within each pixel cluster, is pooled from ALL the edges to determine the omnibus correction criterion.
5.Show pre-selected ROIs
This plot shows the preselected pairwise dipole density (i.e., unweighted graph edges). The connectivity measure is this pairwise dipole density weighted by rPDC or dDTF.
6.View results & Export for movie (08/20/2020 updated)
Specify the *_tStatistics.mat and *_dipolePairDensity.mat files. Enter all the parameters. The ‘Cluster-level correction’ here determines the threshold on mass of cluster. After pressing ‘Plot connectivity matrix’, click a graph edge in the main connectivity matrix plot on the left.
-MCC for graph edges (checkbox)–MCC stands for multiple comparison correction. When checked, surrogate distributions of extreme values (minimum and maximum statistics) will be built using 10,000 values (default) of min/max value across ALL graph edges submitted (including the ones with no significant results). Then, for the case of p < 0.05 (default), the 2.5- and 97.5-percentile values of the surrogate distribution are obtained with which one can perform omnibus correction for time-frequency values of any graph edge at the same time.
If unchecked, surrogate distributions will be built using 10,000 (default) x [number_of_edges] values across all graph edges submitted. This surrogate statistics distribution is NOT made of the max/min values across all graph edges, so the 2.5- and 97.5-percentile values of the surrogate distribution cannot address multiple comparison across graph edges, but it still does for any single graph edge. Thus, unchecking this options should be used with caution, for example for hypothesis-driven ROI analysis.
-Use GFWER (u=1)(checkbox)–GFWER stands for generalized (weak) family-wise error rate. ‘Weak’ means using cluster-level correction.
The trade off you make here is that you gain more detection power at the cost of the fact that you accept maximum 1 (hence u=1) false positive result (in your case, one cluster of pixels) present in your result. This may sound unusual and even scary, but remember that you are already always accepting 5% of false positive results which is usually WAY larger than 1. How it works is as follows. GFWER does not pick up the max/min statistics from each iteration of permutation trial, but the second to max/min (only one next to the max/min, hence u=1). This approach is to gain statistical sensitivity at the cost of known number of false positive results (here, u=1 i.e., one mass of cluster in your data is known to be a result of false positive). You may wonder if this is meaningful thing to do. It is, because a distributions of surrogate statistics tend to have outliers in tails. Removing the leftmost and rightmost values from the tails can in most cases greatly ease the extreme value statistics. You can try to find out how effective this trade could be, as it is calculated altogether anyway. By the way, this option is only usable when ‘MCC for graph edges’ option is checked.
Below, three statistical results are shown from the same data. From left, graph-edge MCC on, graph-edge MCC on with GFWER, and graph-edge MCC off. Note the change of the number of significant edges. Data are from Loo et al. (2019).
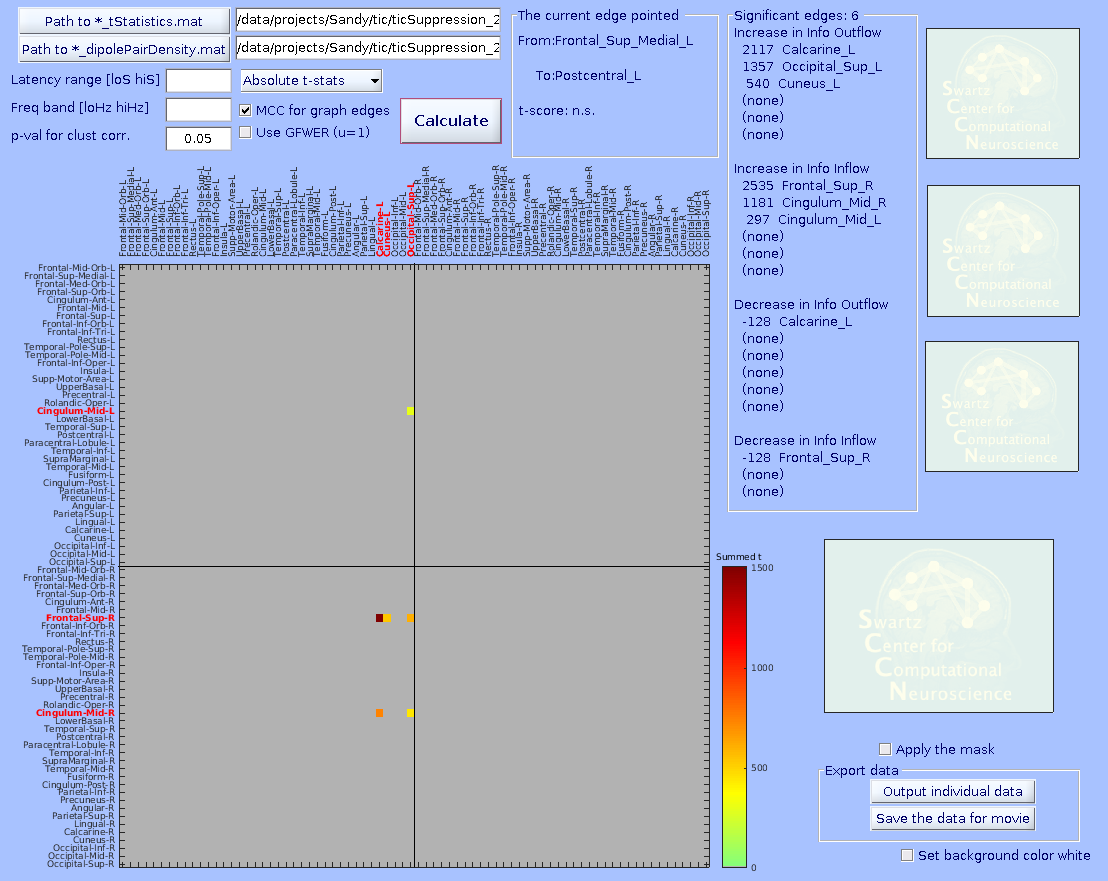
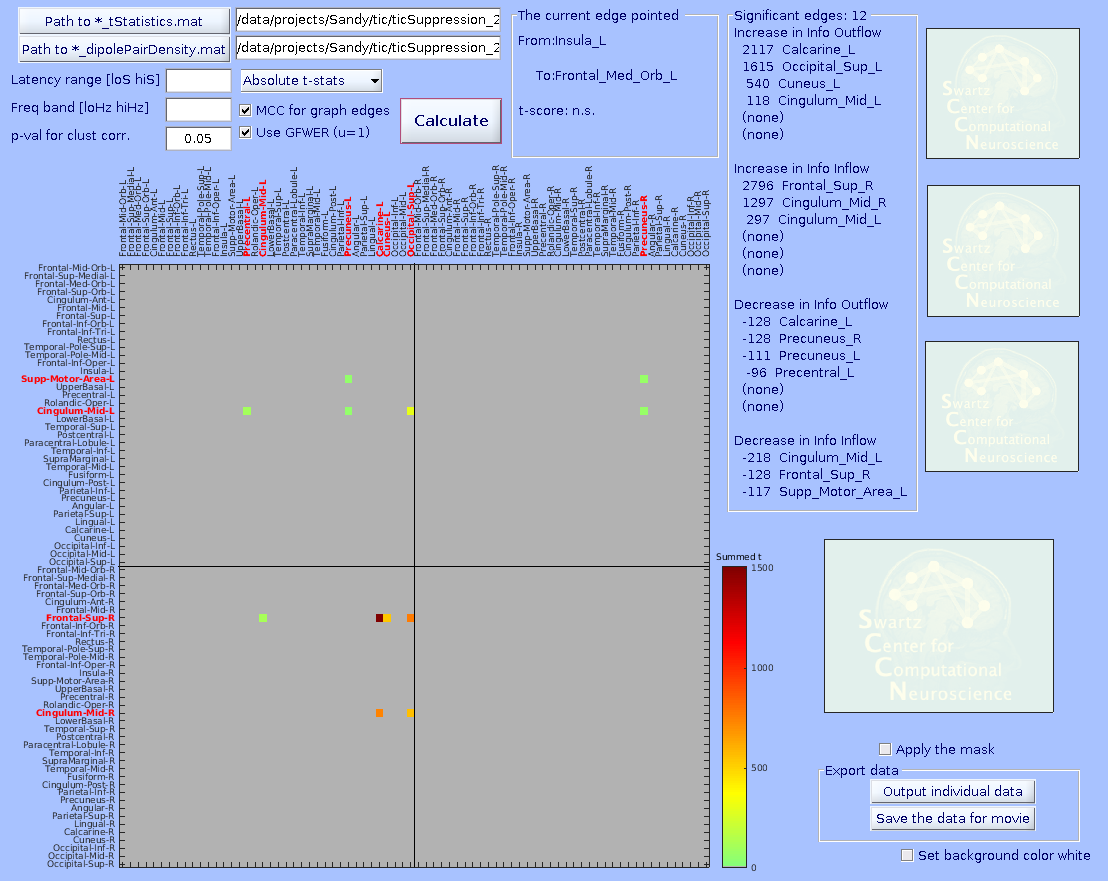
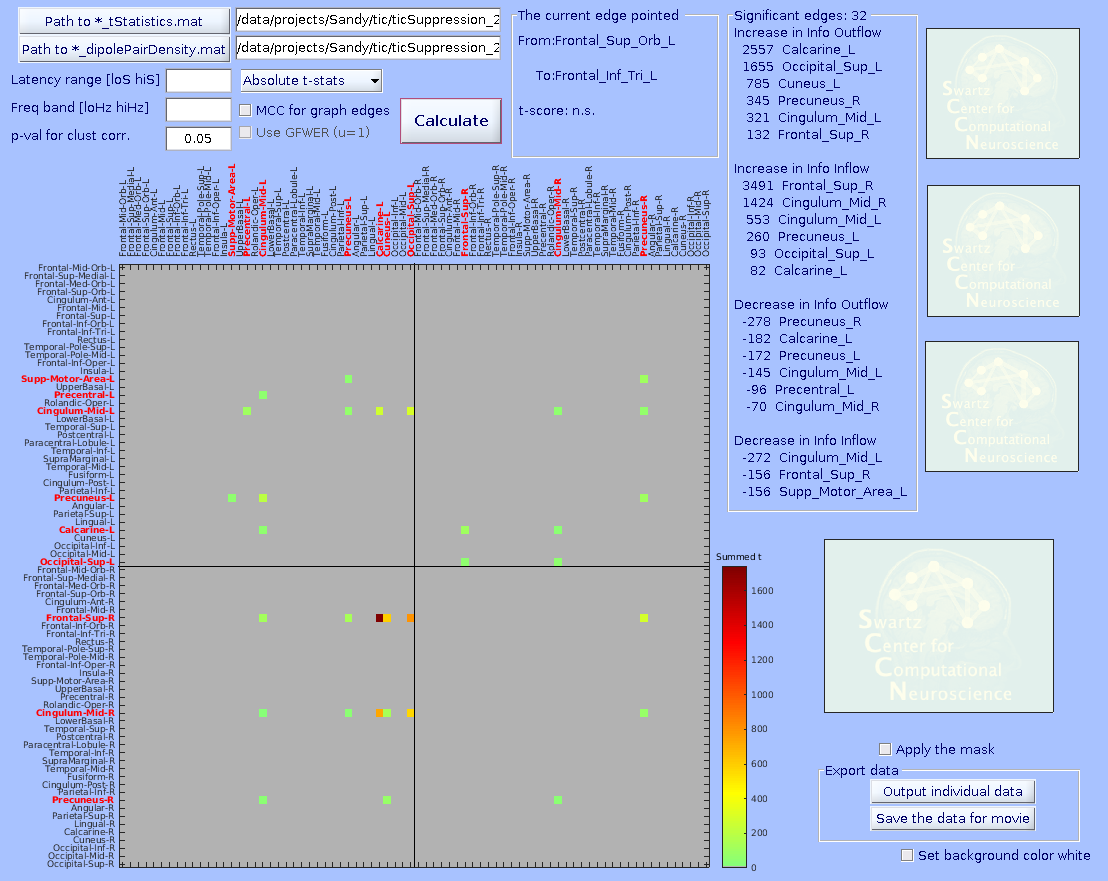
How to generate a group-level connectivity movie
- The button ‘Save the data for movie’ is located at the bottom right corner of the visualization GUI.
- All the parameters determined in View results & Export for movie will take effect on the movie data.
- Provide any one of the .set files used for the analysis. The copy of the specified .set file serves as a ‘donor’, and its EEG.CAT.Conn.RPDC/dDTF08 is replaced with the group-mean values after thresholding.
- From EEGLAB main window, Tools -> SIFT -> Visualiation -> BrainMovie3D. I use my customized movie function for this. This opens the propertyGrid interface, which is known for multiple uses and dependencies. Care must be taken to set Matlab path before using this function. Use ‘which -all propertyGrid’ to ensure you use the right one, otherwise it won’t work.
- “FrequenciesToCollapse” may need to be adjusted so that in my case instead of 2:50 I need to set 2:49.9 to make it work.
- Do not subtract baseline. It is taken care of by groupSIFT. Otherwise, zero values (i.e., masked by statistical results) will be non-zeros.
- “FooterPanelDisplaySpec”, “GraphMetric”. This will show you envelope time-series.
- “InitialView” [50 36] is the default value. For axial slice, [0 90]; for sagittal slice, [90 0]; for coronal slice, [0 0].
- “Theme”, “darkdream” (optional)
- “ImageOutputDirectory”, “prompt” (you need to type it) By the way, “Save all picture frames” currently does not work, but you still need to enter ‘prompt’ twice for picture and movie; one for the path and the other for the file name.
- “MovieOutputFinename”, “prompt” (you need to type it)
How to generate a group-level connectivity movie (12/23/2019 update)
On the ‘pop_viewResultsAndExportForMovie’ GUI, there is a ‘Output individual data’ button on the bottom right. When you click it, uigetdir() GUI pops up and asks you to select ‘_allSubjStack.mat’ file. After selecting this file, it generates singificant blob-by-blob mean value for individual subjects with which one can perform correlation analysis. The output is saved as ‘dataSheet’ on Matlab ‘base’ workspace, which you can export as csv file. For the case of A-B, you should perform for A and B separately (both A and B have the same graph edges).
How to output individual subject data in the case of subtraction (02/26/2020 Updated)
Currently, this is not supported by GUI. But with fairly simple command line operation, you can do it IF THE TWO CONDITIONS ARE WITHIN-SUBJECT.
- Empty your workspace.
- Load XXXX_allSubjStack.mat for Condition A.
- Rename ‘allConnectivityStack’ to something else (here, ‘tmp1’)
- Load YYYY_allSubjStack.mat for Condition B. Note that if A and B are within-subject condition (i.e., same ICA results), they should have the same data except for ‘allConnectivityStack’.
- Rename ‘allConnectivityStack’ to something else (here, ‘tmp2’)
- Perform allConnectivityStack = tmp1-tmp2;
- Save everything in the workspace with a new name ZZZZ_allSubjStack.mat.
- Feed ZZZZ_allSubjStack.mat during GUI operation.
The same method can be used to take subtraction between within-subject conditions. For example, if you have mismatch negativity data for two group of subjects, you want to subtract Deviant-Standard for each group using the method explained above, then perform group-level analysis. Note that in this case, you have to obtain individual data list for each group separately.
Layout issue (06/21/2018 update)
For unknown reason, GUI layout can be collapsed in your environment. Since I can’t replicate the issue in my environment, for the time being I would like the users to fix it themselves following these steps.
- Type ‘guide’ in Matlab command line.
- Top tab ‘Open existing GUI’ and ‘Browse’ button to specify the groupSIFT GUI in question under your eeglab plugin folder.
- ‘Open’ button to open the GUI in question.
- Manually fix the layout and save. I even heard that one of the windows were hidden by another window… so if you don’t see what you are looking for, do not forget to check the background of the things on surface.
Link to the latest workshop material
EEGLAB workshop 2017 in Tokyo Link to a movie example
About the custom anatomical labels
groupSIFT uses anatomical labels defined in Automated Anatomical Labeling solution (Tzourio-Mazoyar et al., 2002). However, instead of the original 88 regions, I reduced it to 76 regions by integrating 16 small regions in limbic and basal regions into umbrella ROIs ‘Upper Basal’ and ‘Lower Basal’. In pop_groupSIFT_convertToGroupAnatomicalRois.m line 307-402, there is the following description.
% These regions are to be included
% 'Precentral_L'
% 'Precentral_R'
% 'Frontal_Sup_L'
% 'Frontal_Sup_R'
% 'Frontal_Sup_Orb_L'
% 'Frontal_Sup_Orb_R'
% 'Frontal_Mid_L'
% 'Frontal_Mid_R'
% 'Frontal_Mid_Orb_L'
% 'Frontal_Mid_Orb_R'
% 'Frontal_Inf_Oper_L'
% 'Frontal_Inf_Oper_R'
% 'Frontal_Inf_Tri_L'
% 'Frontal_Inf_Tri_R'
% 'Frontal_Inf_Orb_L'
% 'Frontal_Inf_Orb_R'
% 'Rolandic_Oper_L'
% 'Rolandic_Oper_R'
% 'Supp_Motor_Area_L'
% 'Supp_Motor_Area_R'
% 'Frontal_Sup_Medial_L'
% 'Frontal_Sup_Medial_R'
% 'Frontal_Med_Orb_L'
% 'Frontal_Med_Orb_R'
% 'Rectus_L'
% 'Rectus_R'
% 'Insula_L'
% 'Insula_R'
% 'Cingulum_Ant_L'
% 'Cingulum_Ant_R'
% 'Cingulum_Mid_L'
% 'Cingulum_Mid_R'
% 'Cingulum_Post_L'
% 'Cingulum_Post_R'
% 'Calcarine_L'
% 'Calcarine_R'
% 'Cuneus_L'
% 'Cuneus_R'
% 'Lingual_L'
% 'Lingual_R'
% 'Occipital_Sup_L'
% 'Occipital_Sup_R'
% 'Occipital_Mid_L'
% 'Occipital_Mid_R'
% 'Occipital_Inf_L'
% 'Occipital_Inf_R'
% 'Fusiform_L'
% 'Fusiform_R'
% 'Postcentral_L'
% 'Postcentral_R'
% 'Parietal_Sup_L'
% 'Parietal_Sup_R'
% 'Parietal_Inf_L'
% 'Parietal_Inf_R'
% 'SupraMarginal_L'
% 'SupraMarginal_R'
% 'Angular_L'
% 'Angular_R'
% 'Precuneus_L'
% 'Precuneus_R'
% 'Paracentral_Lobule_L'
% 'Paracentral_Lobule_R'
% 'Temporal_Sup_L'
% 'Temporal_Sup_R'
% 'Temporal_Pole_Sup_L'
% 'Temporal_Pole_Sup_R'
% 'Temporal_Mid_L'
% 'Temporal_Mid_R'
% 'Temporal_Pole_Mid_L'
% 'Temporal_Pole_Mid_R'
% 'Temporal_Inf_L'
% 'Temporal_Inf_R'
%
% These regions are to be combined
% 'Hippocampus_L'
% 'Hippocampus_R'
% 'ParaHippocampal_L'
% 'ParaHippocampal_R'
% 'Amygdala_L'
% 'Amygdala_R'
% --> Lower Basal
%
% 'Olfactory_L'
% 'Olfactory_R'
% 'Caudate_L'
% 'Caudate_R'
% 'Putamen_L'
% 'Putamen_R'
% 'Pallidum_L'
% 'Pallidum_R'
% 'Thalamus_L'
% 'Thalamus_R'
% --> Upper Basal
%
% One can visualize these regions by running visualizeAnatomicalRoiWithNHimasBlobs.m contained by the groupSIFT folder.
The reason why I created the umbrella ROIs is because these limbic and basal regions (even including ventricles) are unlikely to be generators of scalp-measurable EEG due to lack of critical conditions, namely a large area of pyramidal cells aligned in parallel. However, because of errors in dipole fitting, about 20% of fitted dipoles goes into these physiologically invalid deep retions (for detail, see this page). If we know the label ‘thalamus’ is completely inappropriate to be used to refer to the estimated EEG sources, should we still provide specific labels that are now only misleading? Instead, I suggest that we use umbrella terms ‘Upper Basal’ and ‘Lower Basal’ just to indicate how deep they are. The depth information in dipole fitting could be related to the area information in the actual dipole sheet, so making a minimal distinction between ‘upper’ and ‘lower’ may be helpful.
Published works
The dedicated technical paper is not prepared yet. But a couple of clinical researches using groupSIFT are already published. Loo et al. (2019) has relatively detailed description of the method in Supplement (which needs some update).
Download
GroupSIFT is NOT in the EEGLAB plugin manager. You may install GroupSIFT by downloading the zipped file from the GitHub repository, ‘Code’ (the green button) -> ‘Download ZIP’ (the menu item at the botton). Unzip the file and place the resulting folder in the EEGLAB plugin folder. The current version is 0.51.
Support
groupSIFT was developed for a project for a study on chronic tic disorder (PI Sandra Loo) that was supported by NINDS 80160 and 97484.